Hi,
1. Problem
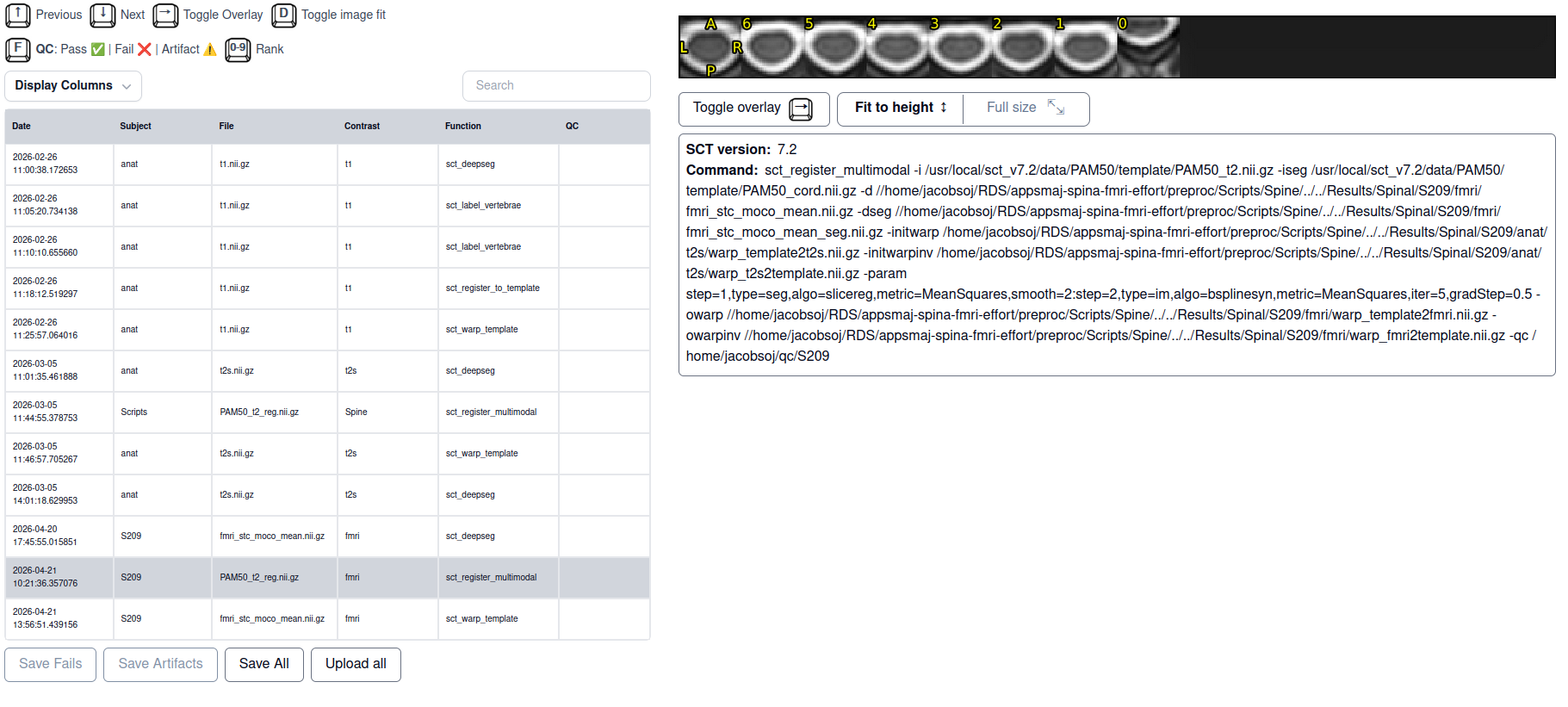
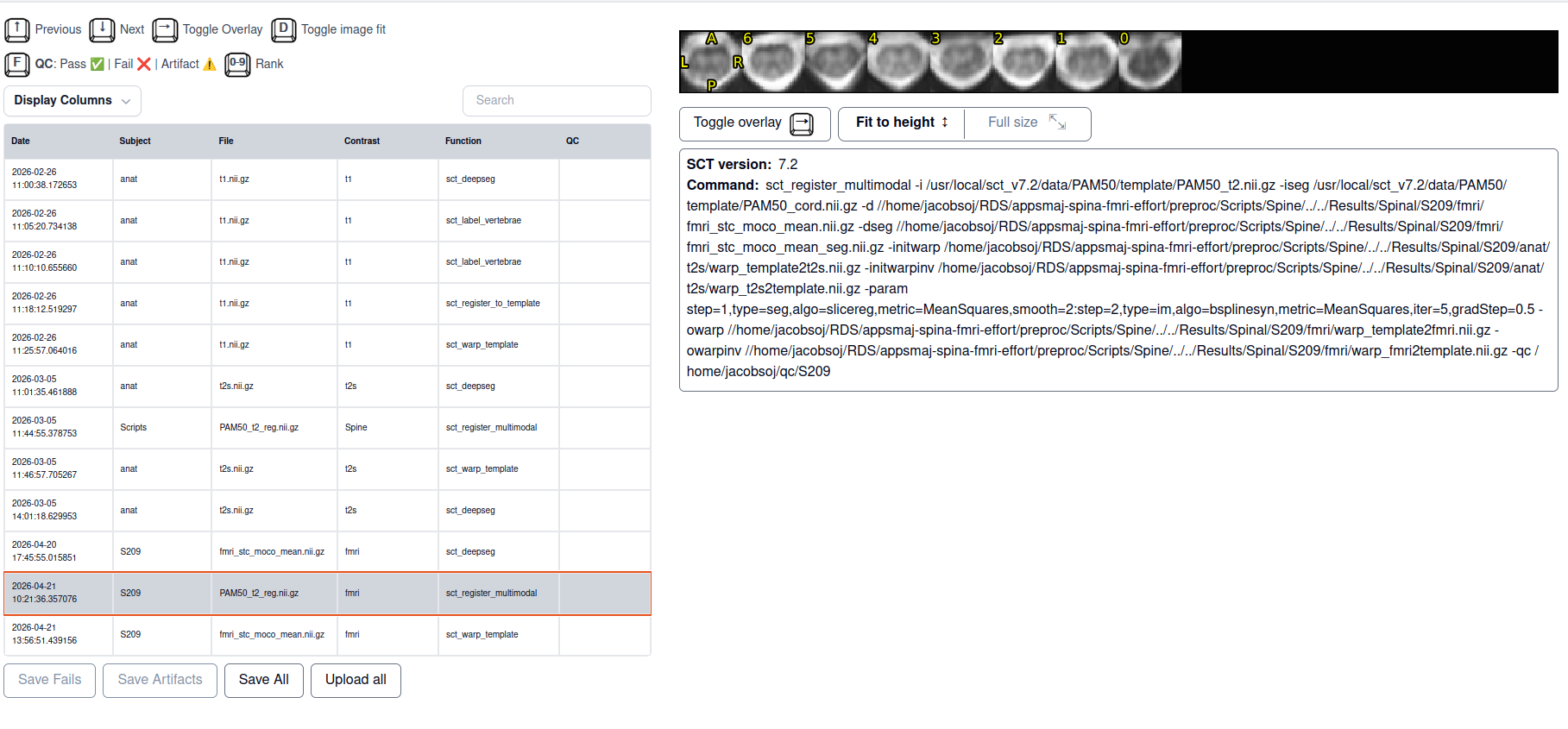
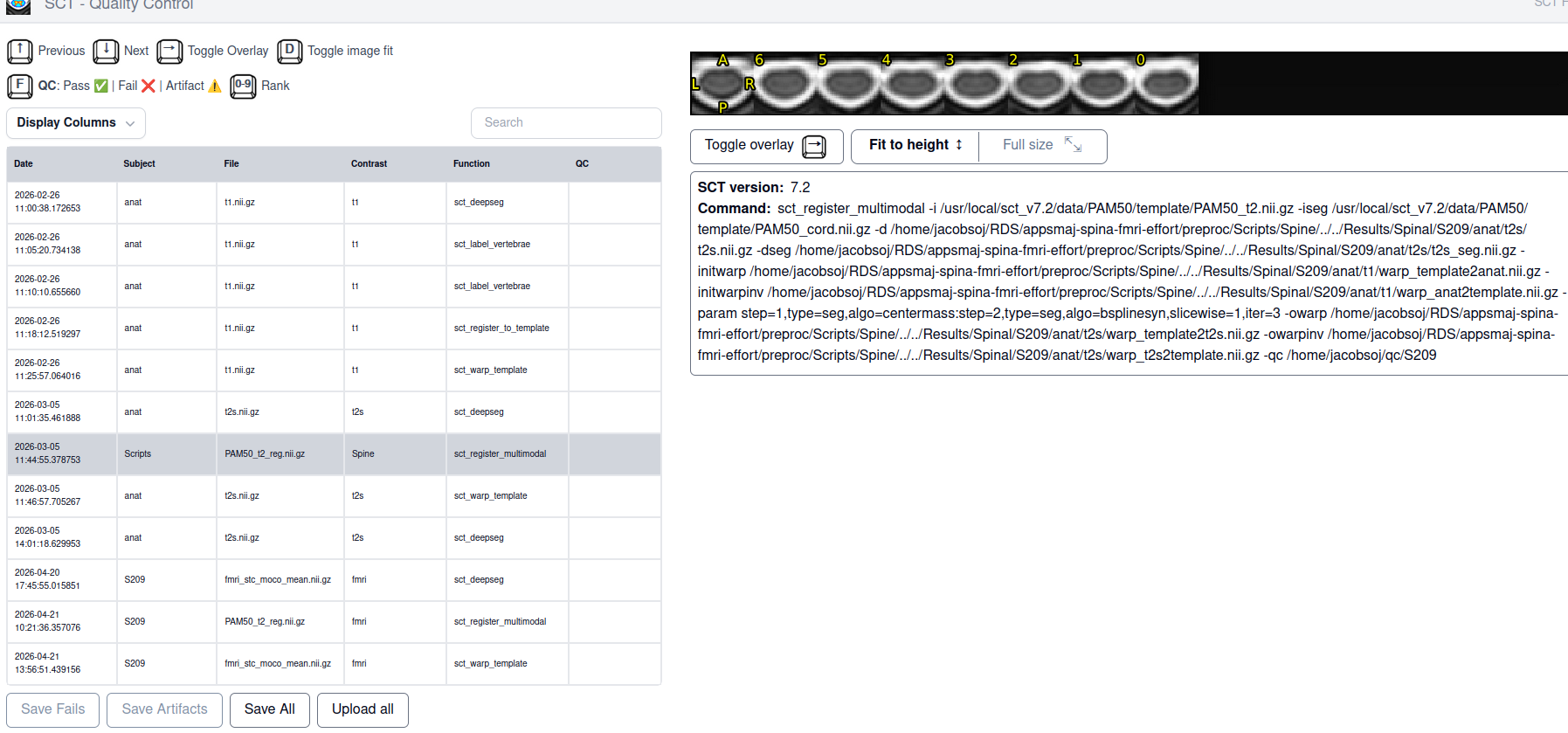
I am trying to register the PAM50 template to my spinal fMRI data. For a couple of subjects that was successful, now I have a subject for who in the registration goes well except for in the lowest slice where there is suddenly a big jump in spatial location. tSNR for that slice is similar to other slices. What would be the correct way of dealing with this?
From README:
Script 5. `batch_processing_fMRI_FDM.sh`
Purpose: Main fMRI preprocessing pipeline. Produces a motion-corrected,
template-registered 4D fMRI volume with PAM50 atlas labels in fMRI space.
Requires: T1 results and T2s results
Steps:
| 1 | Temporal SNR (tSNR) map — quality check before any processing |
| 2 | Remove initial volumes |
| 3 | Ghosting correction — interactive, per-slice circular shift if needed |
| 4 | Slice timing correction (`slicetimer` via FSL, using `SP_slicetiming.txt`) |
| 5 | Motion correction (`sct_fmri_moco` with cord mask; middle volume as starting reference for iterative averaging) |
| 6 | Mean fMRI segmentation (`sct_deepseg sc_epi`) |
| 7 | Template registration: T2s-space template → fMRI (`sct_register_multimodal`), then warp concatenation T1→T2s→fMRI |
| 8 | Atlas warping to fMRI space (`sct_warp_template`) |
2. Commands and terminal output:
step 6 successful, followed by step 7 in which the jump appears:
# Step 6: Segment Mean fMRI Volume
echo "=== Step 6: Segmenting Mean fMRI Volume ==="
# Use the mean from motion-corrected data.
fmri_mean=${out_dir}/fmri_stc_moco_mean.nii.gz
echo "Segmenting mean fMRI volume with deepseg..."
cd ${out_dir}
sct_deepseg sc_epi -i ${fmri_mean} -qc ~/qc/${SUBJECT}
cd ${dir_now}
# sct_deepseg sc_epi appends _bold_seg to the input stem; rename to plain _seg
# for consistency with the rest of the pipeline.
if [ -f "${out_dir}/fmri_stc_moco_mean_bold_seg.nii.gz" ]; then
mv "${out_dir}/fmri_stc_moco_mean_bold_seg.nii.gz" "${out_dir}/fmri_stc_moco_mean_seg.nii.gz"
echo "✓ fMRI segmentation completed"
elif [ -f "${out_dir}/fmri_stc_moco_mean_seg.nii.gz" ]; then
echo "✓ fMRI segmentation completed"
else
echo "ERROR: fMRI segmentation failed"
exit 1
fi
echo ""
[manual checkpoint echo statements]
# Step 7: Register Template to fMRI
echo "=== Step 7: Registering Template to fMRI ==="
echo "Using -initwarp from T2* registration for initialisation"
echo ""
# Run from out_dir so that sct_register_multimodal deposits the registered
# template image (PAM50_t2_reg.nii.gz) there rather than in the launch directory.
cd ${out_dir}
sct_register_multimodal \
-i "${SCT_DIR}/data/PAM50/template/PAM50_t2.nii.gz" \
-iseg "${SCT_DIR}/data/PAM50/template/PAM50_cord.nii.gz" \
-d ${fmri_mean} \
-dseg ${out_dir}/fmri_stc_moco_mean_seg.nii.gz \
-initwarp "${T2s_dir}/warp_template2t2s.nii.gz" \
-initwarpinv "${T2s_dir}/warp_t2s2template.nii.gz" \
-param step=1,type=seg,algo=slicereg,metric=MeanSquares,smooth=2:step=2,type=im,algo=bsplinesyn,metric=MeanSquares,iter=5,gradStep=0.5 \
-owarp ${out_dir}/warp_template2fmri.nii.gz \
-owarpinv ${out_dir}/warp_fmri2template.nii.gz \
-qc ~/qc/${SUBJECT}
cd ${dir_now}
if [ $? -eq 0 ]; then
echo "✓ Template registered to fMRI"
echo " Template → fMRI: ${out_dir}/warp_template2fmri.nii.gz"
echo " fMRI → Template: ${out_dir}/warp_fmri2template.nii.gz"
echo " Registered template image: ${out_dir}/PAM50_t2_reg.nii.gz"
else
echo "ERROR: Template registration failed"
exit 1
fi
echo ""
3. System information
SCT info:
- version: 7.0
- path: /Users/selmalugtmeijer/sct_7.0
OS: osx (macOS-15.7.4-x86_64-i386-64bit)
CPU cores: Available: 8, Used by ITK functions: 8
RAM: Total: 24576MB, Used: 2823MB, Available: 2148MB
OPTIONAL DEPENDENCIES
---------------------
Check FSLeyes version...............................[OK] (1.14.2)
MANDATORY DEPENDENCIES
Check Python executable.............................[OK]
Using bundled python 3.9.23 | packaged by conda-forge | (main, Jun 4 2025, 18:00:50)
[Clang 18.1.8 ] at /Users/selmalugtmeijer/sct_7.0/python/envs/venv_sct/bin/python3.9
Check if acvl_utils is installed....................[OK]
Check if dipy is installed..........................[OK] (1.8.0)
Check if ivadomed is installed......................[OK] (2.9.10)
Check if matplotlib is installed....................[OK] (3.9.4)
Check if matplotlib-inline is installed.............[OK]
Check if monai is installed.........................[OK] (1.4.0)
Check if nibabel is installed.......................[OK] (5.3.2)
Check if nilearn is installed.......................[OK] (0.10.4)
Check if nnunetv2 is installed......................[OK]
Check if numpy is installed.........................[OK] (1.26.4)
Check if onnxruntime is installed...................[OK] (1.19.2)
Check if pandas is installed........................[OK] (1.5.3)
Check if portalocker is installed...................[OK] (3.1.1)
Check if psutil is installed........................[OK] (7.0.0)
Check if pyqt5 (5.12.3) is installed................[OK] (5.12.3)
Check if pyqt5-sip is installed.....................[OK]
Check if pystrum is installed.......................[OK] (0.4)
Check if pytest is installed........................[OK] (8.3.5)
Check if pytest-cov is installed....................[OK] (6.1.1)
Check if requests is installed......................[OK] (2.32.3)
Check if requirements-parser is installed...........[OK] (0.11.0)
Check if scipy is installed.........................[OK] (1.13.1)
Check if scikit-image is installed..................[OK] (0.24.0)
Check if scikit-learn is installed..................[OK] (1.6.1)
Check if totalspineseg is installed.................[OK] (20250205)
Check if xlwt is installed..........................[OK] (1.3.0)
Check if torch is installed.........................[OK] (2.2.2)
Check if tqdm is installed..........................[OK] (4.67.1)
Check if transforms3d is installed..................[OK] (0.4.2)
Check if urllib3 is installed.......................[OK] (2.4.0)
Check if pytest_console_scripts is installed........[OK]
Check if pyyaml is installed........................[OK] (6.0.2)
Check if voxelmorph is installed....................[OK] (0.2)
Check if wquantiles is installed....................[OK] (0.4)
Check if xlsxwriter is installed....................[OK] (3.2.3)
Check if spinalcordtoolbox is installed.............[OK]
Check ANTs compatibility with OS ...................[OK]
Check PropSeg compatibility with OS ................[OK]
Check if figure can be opened with PyQt.............[OK]
Check if figure can be opened with matplotlib.......[OK] (Using GUI backend: 'qtagg')
Check data dependency 'PAM50'.......................[OK]
Check data dependency 'deepseg_gm_models'...........[OK]
Check data dependency 'deepseg_sc_models'...........[OK]
Check data dependency 'deepseg_lesion_models'.......[OK]
Check data dependency 'deepreg_models'..............[OK]
Check data dependency 'PAM50_normalized_metrics'....[OK]
Check data dependency 'binaries_osx'................[OK]
4. File uploads
- spinal cord segmentation on the fMRI image which looks good
- template registration with big jump only in the lowest slice
- SCT html QC output for multimodal with template
- SCT html QC output for multimodal without template
- SCT html QC output T2 with template
Uploading: spinal_cord_seg.png…
Uploading: spinal_template_reg.png…