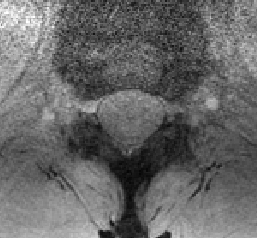
Sorry for not attaching the image to the previous message. Attached is the t2s to determine if gray and white matter CSA can be pulled out.
t2s.nii.gz (3.4 MB)
I am also trying to pull MTR from the below files, but when I get to the point of putting the values into an Excel sheet the values are similar between white matter and the corticospinal tracts.
mt1.nii.gz (1.3 MB)
mt0.nii.gz (1.3 MB)
Get centerline from mt1 data
sct_get_centerline -i mt1.nii.gz -c t2
Create mask
sct_create_mask -i mt1.nii.gz -p centerline,mt1_centerline.nii.gz -size 45mm
Crop data for faster processing
sct_crop_image -i mt1.nii.gz -m mask_mt1.nii.gz -o mt1_crop.nii.gz
Segment spinal cord
sct_deepseg_sc -i mt1_crop.nii.gz -c t2 -qc qc
Register mt0->mt1
sct_register_multimodal -i mt0.nii.gz -d mt1_crop.nii.gz -dseg mt1_crop_seg.nii.gz -param step=1,type=im,algo=rigid,slicewise=1,metric=CC -x spline -qc qc
Register template->mt1
sct_register_multimodal -i $SCT_DIR/data/PAM50/template/PAM50_t2.nii.gz -iseg $SCT_DIR/data/PAM50/template/PAM50_cord.nii.gz -d mt1_crop.nii.gz -dseg mt1_crop_seg.nii.gz -param step=1,type=seg,algo=slicereg,smooth=3:step=2,type=seg,algo=bsplinesyn,slicewise=1,iter=3 -initwarp …/t2/warp_template2anat.nii.gz -initwarpinv …/t2/warp_anat2template.nii.gz
Rename warping fields for clarity
mv warp_PAM50_t22mt1_crop.nii.gz warp_template2mt.nii.gz
mv warp_mt1_crop2PAM50_t2.nii.gz warp_mt2template.nii.gz
Warp template
sct_warp_template -d mt1_crop.nii.gz -w warp_template2mt.nii.gz -qc qc
Compute mtr
sct_compute_mtr -mt0 mt0_reg.nii.gz -mt1 mt1_crop.nii.gz
Extract MTR
sct_extract_metric -i mtr.nii.gz -method map -l 51 -o mtr_in_wm.csv
sct_extract_metric -i mtr.nii.gz -method map -l 52 -o mtr_in_gm.csv
sct_extract_metric -i mtr.nii.gz -method map -l 4 -o mtr_in_left_lateral_corticospinal_tract.csv
sct_extract_metric -i mtr.nii.gz -method map -l 5 -o mtr_in_right_lateral_corticospinal_tract.csv